
Research Overview
1.AIEnhanced Molecular Simulation Techniques
2.Understanding Anomalous Thermal Transport in amorphous materials
3.Simulation of Soft Porous Materials


Latest Works
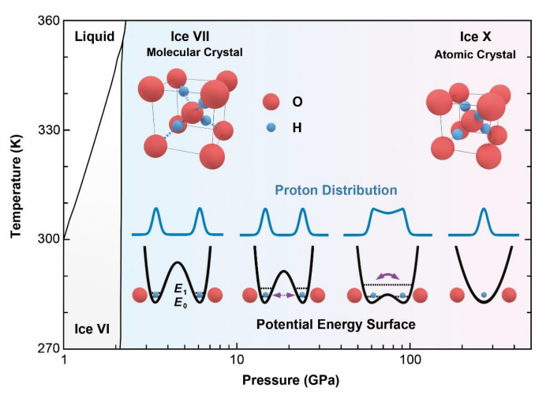
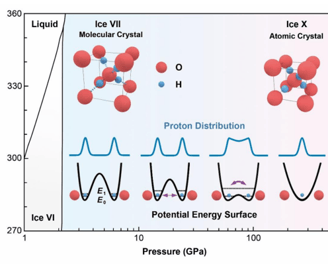
Proton tunneling is believed to be nonlocal in ice, but its range has been shown to be limited to only a few molecules. Here, we measured the thermal conductivity of ice under pressure up to 50 GPa and found it increases with pressure until 20 GPa but decreases at higher pressures. We attribute this nonmonotonic thermal conductivity to the collective tunneling of protons at high pressures, supported by large-scale quantum molecular dynamics simulations. The collective tunneling loops span several picoseconds in time and are as large as nanometers in space, which match the phonon periods and wavelengths, leading to strong phonon scattering at high pressures. Our results show direct evidence of global quantum motion existing in high-pressure ice and provide a new perspective to understanding the coupling between phonon propagation and atomic tunneling.
Proton Collective Quantum Tunneling Induces Anomalous Thermal Conductivity of Ice under Pressure
Wang, Yufeng, et al. "Proton Collective Quantum Tunneling Induces Anomalous Thermal Conductivity of Ice under Pressure." Physical Review Letters 132.26 (2024): 264101.
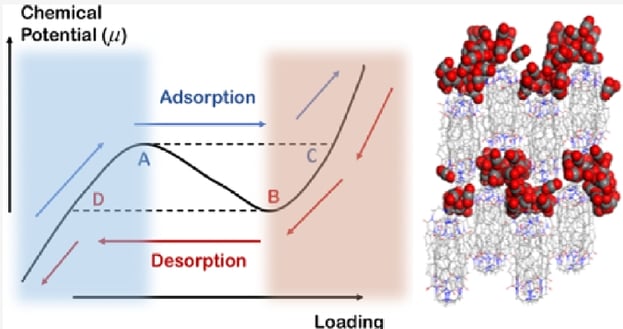
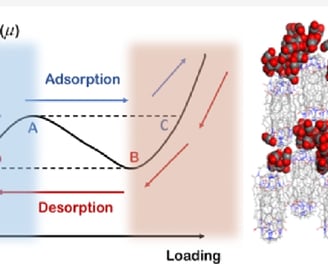
Soft porous crystals (SPCs) are novel porous materials that exhibit significant global crystal structure deformations under external stimuli. The crystal structure deformation brings strong switch effects and hysteresis effects, which can be used to achieve highly selective separations for similar gas molecules. Among SPCs, soft porous organic cages (POCs) are particularly interesting due to their unique advantages in stability and processability. The soft nonbonding interactions in POCs lead to more delicate structural deformations and complicated sorption isotherms. Therefore, compared to rigid porous materials, it is more difficult to understand, predict, and rationally control the gas uptakes of POCs. In this work, we advance this field by employing a hybrid Grand Canonical Monte Carlo/Molecular Dynamics strategy to investigate the unique sorption behavior of a POC, named NKPOC-1, upon exposure to CO2. The experimental multistep sorption isotherms with large hysteresis at different temperatures are well reproduced. We further elucidate the distinct sorption and crystal structure deformation mechanisms at different temperatures, demonstrating the unusual complexity of POC gas adsorption. To the best of our knowledge, this is the first thorough theoretical study of understanding the sorption mechanism of soft POCs. Also, the methodology used in this work provides a generally effective way to simulate the adsorption behaviors in highly flexible materials.
Molecular Dynamics Simulation of CO2 Uptake in NKPOC-1: Understanding the Temperature-Dependent Multistep Adsorption in Soft Porous Molecular Crystal
Yang, Lan, et al. "Molecular Dynamics Simulation of CO2 Uptake in NKPOC-1: Understanding the Temperature-Dependent Multistep Adsorption in Soft Porous Molecular Crystal." The Journal of Physical Chemistry C 128.26 (2024): 10997-11005.
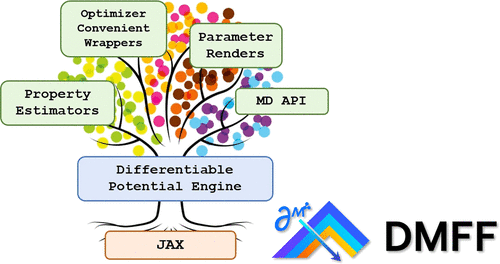
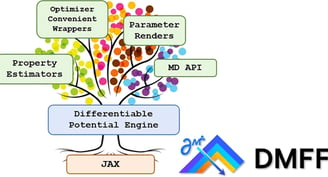
In the simulation of molecular systems, the underlying force field (FF) model plays an extremely important role in determining the reliability of the simulation. However, the quality of the state-of-the-art molecular force fields is still unsatisfactory in many cases, and the FF parameterization process largely relies on human experience, which is not scalable. To address this issue, we introduce DMFF, an open-source molecular FF development platform based on an automatic differentiation technique. DMFF serves as a powerful tool for both top-down and bottom-up FF development. Using DMFF, both energies/forces and thermodynamic quantities such as ensemble averages and free energies can be evaluated in a differentiable way, realizing an automatic, yet highly flexible FF optimization workflow. DMFF also eases the evaluation of forces and virial tensors for complicated advanced FFs, helping the fast validation of new models in molecular dynamics simulation. DMFF has been released as an open-source package under the LGPL-3.0 license and is available at https://github.com/deepmodeling/DMFF.
DMFF: An Open-Source Automatic Differentiable Platform for Molecular Force Field Development and Molecular Dynamics Simulation
Wang, Xinyan, et al. "Dmff: an open-source automatic differentiable platform for molecular force field development and molecular dynamics simulation." Journal of Chemical Theory and Computation 19.17 (2023): 5897-5909.
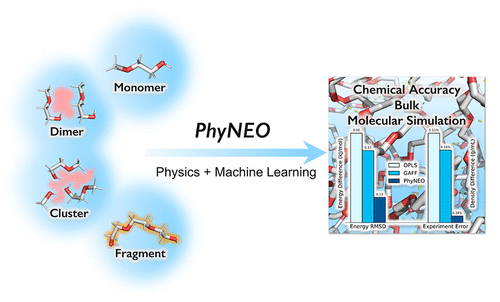
An accurate, generalizable, and transferable force field plays a crucial role in the molecular dynamics simulations of organic polymers and biomolecules. Conventional empirical force fields often fail to capture precise intermolecular interactions due to their negligence of important physics, such as polarization, charge penetration, many-body dispersion, etc. Moreover, the parameterization of these force fields relies heavily on top-down fittings, limiting their transferabilities to new systems where the experimental data are often unavailable. To address these challenges, we introduce a general and fully ab initio force field construction strategy, named PhyNEO. It features a hybrid approach that combines both the physics-driven and the data-driven methods and is able to generate a bulk potential with chemical accuracy using only quantum chemistry data of very small clusters. Careful separations of long-/short-range interactions and nonbonding/bonding interactions are the key to the success of PhyNEO. By such a strategy, we mitigate the limitations of pure data-driven methods in long-range interactions, thus largely increasing the data efficiency and the scalability of machine learning models. The new approach is thoroughly tested on poly(ethylene oxide) and polyethylene glycol systems, giving superior accuracies in both microscopic and bulk properties compared to conventional force fields. This work thus offers a promising framework for the development of advanced force fields in a wide range of organic molecular systems.
PhyNEO: A Neural-Network-Enhanced Physics-Driven Force Field Development Workflow for Bulk Organic Molecule and Polymer Simulations
Chen, Junmin, and Kuang Yu. "PhyNEO: A Neural-Network-Enhanced Physics-Driven Force Field Development Workflow for Bulk Organic Molecule and Polymer Simulations." Journal of Chemical Theory and Computation 20.1 (2023): 253-265.


Research
Dedicated to high-accuracy force field development.
Innovation
contact@yukuanglab.com
+86-0755-36881747
© 2024. All rights reserved.